Ein link zu einem Brief für die Eltern von Dr. Beckwith über BWS :https://www.bwssupport.com/about-us/
(Klick hier wenn du ein bisschen Mut und Positives brauchst 🙂
Hier ist ein Power Point das BWS erklärt:
(Bitte lese dazu unbedingt auch die Infos unter „Screening Protokoll“ auf dieser Website.)
Für mehr Infos über die Genetische Aspekte:eggermann hannover bws treffen 2018_frei(1).
Das BWS -Beckwith Wiedemann Syndrom, auch Exomphalos Makroglossie Gigantismus Syndrom (EMG) genannt, ist ein genetisch bedingtes Grosswuchssyndrom das mit Fehlbildungen und Tumoren verbunden und auf eine Genmutation zurückzuführen ist.
Es wurde von Dr. John Bruce Beckwith (1933) 1964 in Waschington „entdeckt“. Gleichzeitig machte auch Dr. Hans Rudolf Wiedemann (1915) in Kiel die selbe „Entdeckung“.
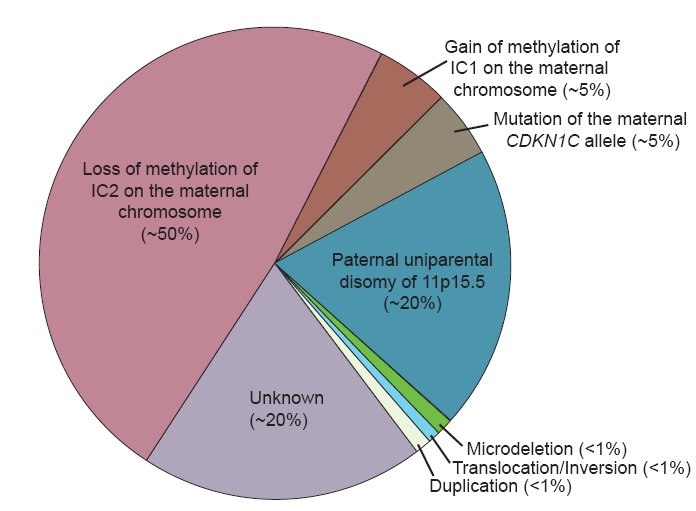
Es gibt verschiedene BWS Typen:
Hier ist eine Diplom arbeit über BWS ( ist schon etwas älter…)
Es folgt eine ausführlichere Zusammenfassung aus orpha.net (dez.2011):
„Das Beckwith-Wiedemann-Syndrom (BWS) ist eine genetische Erkrankung mit Großwuchs, Tumor-Prädisposition und angeborenen Fehlbildungen.
Die Krankheit tritt in allen Ethnien auf, ihre Prävalenz wird auf 1:13.700 geschätzt. Sie ist in beiden Geschlechtern gleich häufig, nur bei eineiigen Zwillingen überwiegt das weibliche Geschlecht.
In der zweiten Hälfte der Schwangerschaft und in den ersten Lebensjahren wachsen die Patienten beschleunigt, während die typische Erwachsenengröße im Normalbereich liegt.
Mögliche Zeichen abnormen Wachstums sind auch Hemihyperplasie und/oder Makroglossie (die zu Schwierigkeiten beim Füttern und Sprechen und, seltener, zu Schlafapnoe führt).
Bei 30-50 % der Neugeborenen besteht eine Hypoglykämie.
Häufig bestehen erkennbare Gesichtszüge, die sich im Erwachsenenalter oft normalisieren.
Charakteristische Befunde sind, neben Makrosomie, Makroglossie, Hemihyperplasie und Hypoglykämie, Omphalozele/Nabelhernie/Rektusdiastase, embryonaler Tumor, Furche(n) an der Vorderseite der Ohrläppchen und Grübchen an der hinteren Helix, Naevus flammeus oder andere Gefäßfehlbildungen, Viszeromegalie der Bauchorgane, fetale adrenokortikale Zytomegalie (pathognomonisch), Nierenfehlbildungen, positive Familienanamnese und, selten, Gaumenspalte. Herzfehler werden bei 9-34 % der Fälle gefunden, und etwa die Hälfte von diesen hat eine sich spontan zurückbildende Kardiomegalie. Eine Kardiomyopathie ist selten.
Die Patienten haben ein hohes Risiko für das Auftreten bösartiger embryonaler Tumoren, besonders in den ersten 8 Lebensjahren, wenn das Risiko 7,5 % (Bereich 4-21 %) beträgt.
Ursache des BWS sind verschiedene epigenetische und/oder genetische Veränderungen, mit gestörter Regulation von genomisch geprägten (imprint) Genen auf Chromosom 11p15.5. Die Krankheit tritt sporadisch in 85 % und familiär in 15 % auf. Molekulare Untergruppen sind mit unterschiedlichen Wiederholungsrisiken und unterschiedlichen klinischen Befunden verbunden.
In der Regel wird die Diagnose durch das Vorhandensein von mindestens drei charakteristischen klinischen Befunden gestützt, aber embryonale Tumoren treten auch bei milden Verläufen auf. Positive molekulare Befunde können die Diagnose bestätigen; negative Ergebnisse schließen aber ein BWS nicht aus.
Differentialdiagnosen sind Simpson-Golabi-Behmel-, Costello-, Perlman- und Sotos-Syndrom und die MPS Typ VI.
Eine vorgeburtliche Diagnostik durch Chorionzottenbiopsie oder Amniozentese kann angeboten werden, besonders wenn eine zytogenetische oder genomische Veränderung identifiziert wurde. Veränderungen der Methylierung sind z. Zt. verlässlicher nach Amniozentese nachzuweisen. Eine Amniozentese ist eventuell auch indiziert, wenn im fetalen Ultraschall BWS-assoziierte Befunde (z. B. fetale Omphalozele) erhoben wurden. Wenn der molekulare Defekt nicht bekannt ist, dienen die Messung des Alpha-Feto-Proteins im mütterlichen Serum und die gezielte Ultraschalluntersuchung als Suchtest.
Die Patienten werden standardgemäß mit unterstützend medizinischen und chirurgischen Verfahren versorgt. Wenn ein BWS vermutet/diagnostiziert wird, soll mit der Tumorüberwachung begonnen werden, auch bei einem klinisch nicht betroffenen eineiigen Zwilling eines Patienten.
Zu diesem Zeitpunkt sollen Genotyp/Phänotyp-Korrelationen für diesen Zweck unberücksichtigt bleiben. Bei verdächtigen oder diagnostischen pränatalen Befunden soll in der Neugeborenenzeit regelmäßig auf mögliche Hypoglykämie untersucht werden. In gleicher Weise soll bei klinisch nicht betroffenen Neugeborenen verfahren werden, die nach der Familienanamnese ein erhöhtes Risiko tragen.
Am schweren Ende des klinischen Spektrums tragen die Patienten das Risiko für einen frühen Tod im Gefolge der Komplikationen, die aus Hypoglykämie, Frühgeburtlichkeit, Kardiomyopathie, Makroglossie oder Tumoren resultieren. Patienten, die das Kindesalter überleben, haben in der Regel eine gute Prognose.“